
简介
(European Union Validation 2)是欧盟针对人用药上市许可申请的一套审评标准。以下是对euV2人用药上市许可申请通知和监管指南的简介:
1. 药品注册申请的变更管理:在药品研发和上市后,变更管理是关键环节,涉及临床试验期间的变更和上市后的变更。这些变更需要根据对药品质量、安全性和有效性的影响程度进行分类和申报 。
2. 集中审批程序:对于某些创新药品,如生物制品和治疗特定疾病的药物,必须通过集中审批程序进行申请。这一程序由EMA的CHMP进行科学评审,并向欧盟委员会提出上市建议。一旦批准,在所有欧盟成员国中有效 。
3. 国家程序:大多数药品通过成员国的国家药监机构(NCAs)获得批准。这包括集中程序不适用的创新产品、仿制药和非处方药 。
4. 非集中程序和互认程:非集中程序允许申请人同时向多个欧盟成员国递交上市申请,而互认程序则是在某一成员国已批准的药品申请在其他成员国获得认可 。
5. 药品上市后变更管理:EMA将药品上市后变更分为不同类别,并要求申请人根据变更的风险程度和类型提交相应的申请。这包括需批准的变更、变更前至少30天提交的补充申请、以及可立即实施的微小变更 。
6. 药物警戒:欧盟要求药品上市许可持有人(MAH)负责上市后的药物警戒,并定期提交安全性更新报告。MAH应建立药物警戒体系,包括记录所有疑似药品不良反应,并在规定时间内上报 。
7. 监管科学:EMA在监管科学方面推动了一系列战略目标,包括促进科学与技术在药物研发中的融合、推动协同证据生成、与医疗保健系统合作等,以提高监管决策的科学质量 。
8. 国际认可程序:英国MHRA推出了国际认可程序(IRP),以简化对已通过欧盟及其他指定国家审批的药品的审评流程,该程序自2024年1月1日起实施 。
访问入口
操作介绍
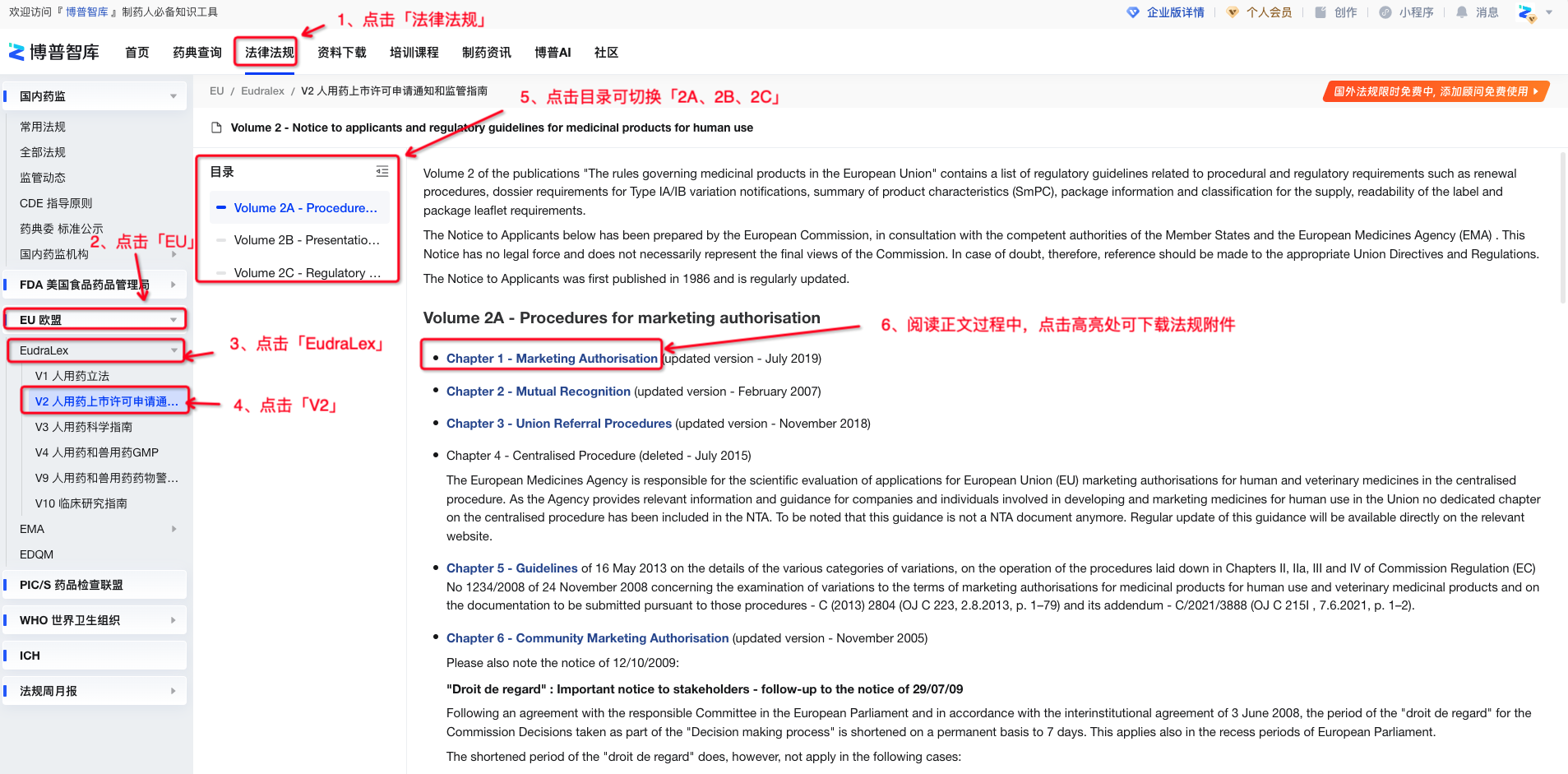
图1-V2
